3D structure determination of macromolecular complexes from heterogeneous samples is still a
major challenge. When variations are highly localized in an otherwise stable structure and can be
identified by 3D variance analysis, a combination of 3D variance analysis with multi-reference
alignment and angular reconstitution can be applied. Often, however, variations involve more
global structural changes and structural variations of flexible macromolecules are often continuous.
The most reliable techniques for analyzing heterogeneous data are electron tomography (ET)
and Random Conical Tilting (RCT). In ET individual 3D structures are first reconstructed and
analyzed for similarity before 3D averaging, while multivariate statistical analysis (MSA) methods
(e.g., correspondence analysis, etc.) are inherent in RCT for differentiating variations in the 0°
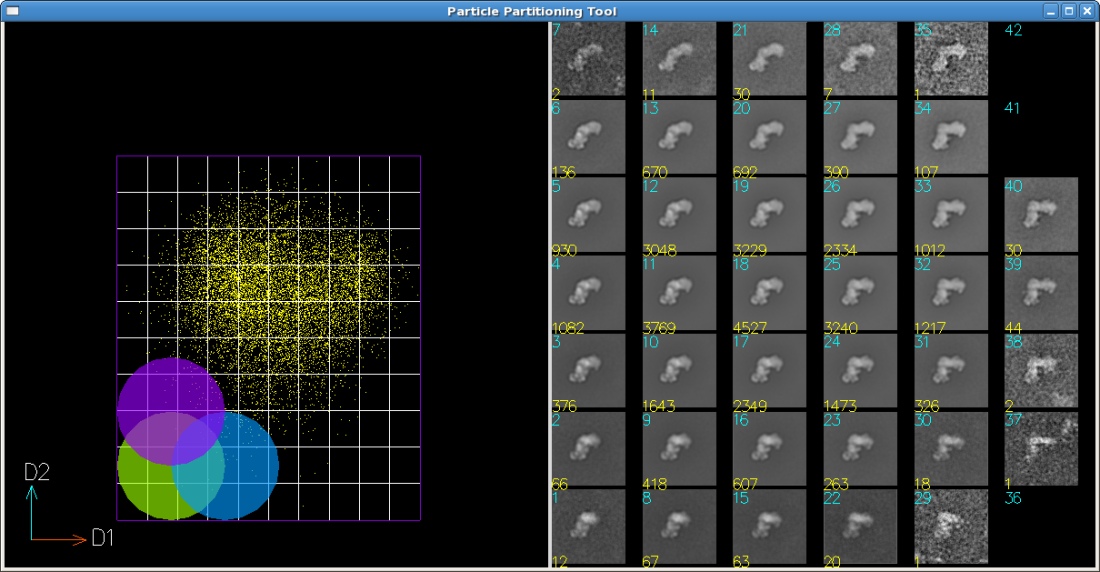
projections of tilt pairs of random conical data. The image data form point clouds in the vector space
generated by the MSA techniques. Traditionally, classification algorithms are used to classify the
data into classes of most identical images. However, classification creates non-overlapping partitions
and can create artificial classes when the variations are continuous. To understand the variations for
each subdivision 3D reconstructions need to be calculated. This requires multiple trials and is
extremely time consuming without dedicated software that automatically keeps track of all
subdivisions and calculates all 3D reconstructions for every new subdivision.
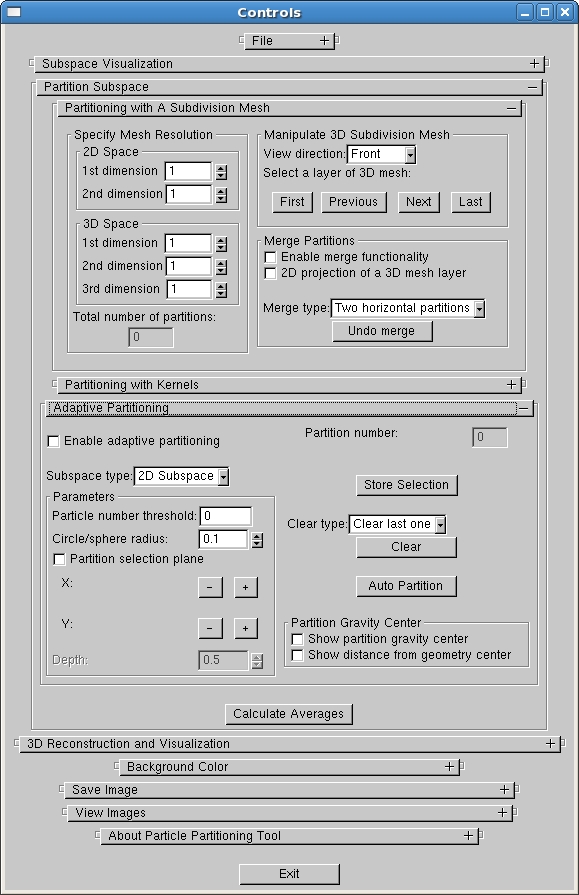
We have developed a software package, HEx3DRM, for the systematic subdivision of image data
based on the image representation in the low-dimensional vector subspace spanned by the first eight
to twelve eigenvectors obtained by MSA methods. The software allows to interactively
subdivide the continuous point cloud using a 2D or 3D subdivision mesh with a user-specified
spacing. In order to generate further meaningful 3D reconstructions, the software allows the
interactive selection of subdivisions which contain few particles and to merge them into larger ones
which contain more particles than a given threshold. This process yields merged subdivisions that
have areas of different polygonal shapes with possibly unequal edge lengths from the initially evenly
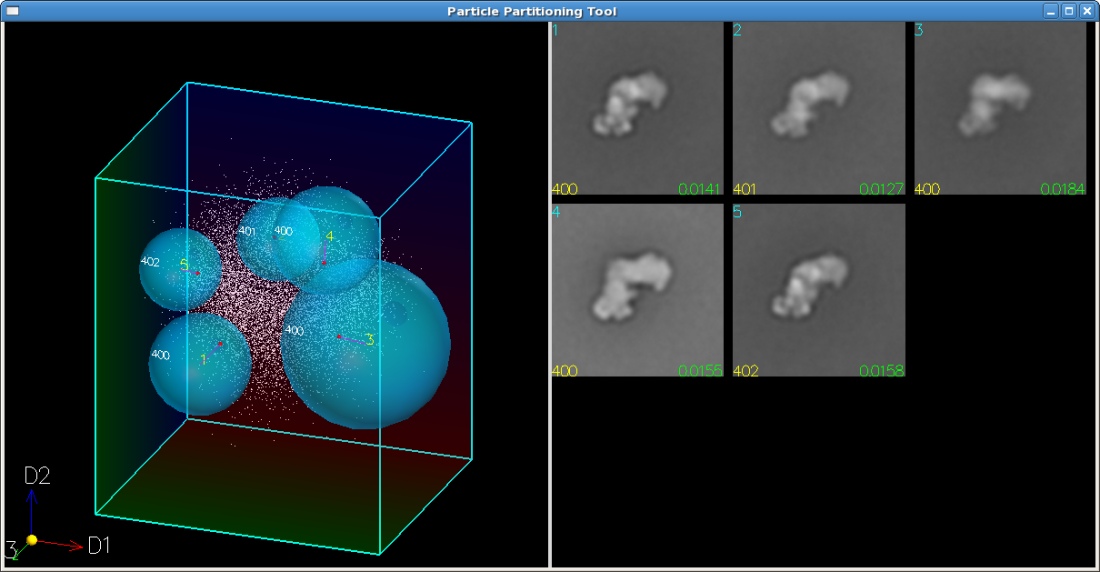
divided subspace. In addition to subdivision based on a regular mesh, kernels of different shapes and
sizes can be used to group particles within a specific geometry and to generate overlapping subsets.
The coordinates of a single particle image may fall into multiple kernel areas, enabling us to analyze
continuous variations in the data set. The software also provides a semi-automatic adaptive
subdivision tool which allows to specify subdivisions at the user-interested positions in the 2D or 3D
subspace. The consistency and quality of each subdivision is assessed by the number of particles in
it. It is planned to combine this with resolution measurements of the corresponding 2D averages.
Based on both measurements, the subdivision size will be adapted such that an optimum balance
between the number of particles and particle variability is achieved. In the automatic 3D
reconstruction process, based on a list of particle membership in each subdivision, a two-step Radon
inversion algorithm calculates the 3D structure for each subdivision from the correspondingly
grouped tilt images. The different macromolecular structures thus can be compared to gain a
better understanding of their conformational variability.
The new tool allows us to browse through the data set, apply a variety of subdivision techniques and
immediately see the result in 3D. This tool is a major step forward to understanding the
structural variations in samples of heterogeneous macromolecules.
Papers:
Yu Zhang, Teresa Ruiz and Michael Radermacher. "HEx3DRM: A heterogeneity explorer for 3D reconstruction of macromolecular complexes". Proc. Microscopy & Microanalysis (M&M2012), pp. 122-123, Phoenix, USA, July 2012.
Copyright 2005-2014, Yu
Zhang. This material may not be published, modified or otherwise
redistributed in whole or part without prior approval.